
En los últimos 60 años, los requisitos para el registro de medicamentos han sufrido importantes cambios aumentando también el control por parte de las Autoridades Sanitarias.
Introducción
La Talidomida, indicada para las náuseas en embarazadas y retirada del mercado en 1961, provocó durante los años 50 y 60, numerosas malformaciones en las extremidades de casi 10.000 fetos. La compañía alegó que no se pudieron detectar estos efectos secundarios antes de la puesta en el mercado del producto.

Por ello, en los últimos 60 años, los requisitos para el registro de medicamentos han sufrido importantes cambios.
Fueron hechos, como el ocurrido con la Talidomida, los que pusieron de manifiesto la necesidad de incrementar los controles en los medicamentos antes de su uso en los pacientes, y no solo de incrementar estos controles, sino de la evaluación, por parte de las autoridades sanitarias, de los resultados obtenidos.
Evolución de la legislación
En el año 1986 se publicó la primera edición del Notice To Applicant (Volumen 2, The Rules governing medicinal Products in the European Union). Tres años más tarde, en 1989, se publicó una actualización completa y los procedimientos europeos, reconocimiento mutuo y centralizado, se aplicaron en 1995.
Más adelante se vio la necesidad de separar la información sobre los procedimientos, de la información sobre el formato y presentación del dosier. Se publicó entonces, en 1998 y de forma separada, el Volumen 2B.
Este volumen incluía guías para la compilación de los dosieres en solicitudes europeas (procedimientos centralizados y de reconocimientos mutuo) y solicitudes nacionales.
De acuerdo a esta legislación, los dosieres debían estructurarse en cuatro partes: parte I (datos administrativos), parte II (documentación de calidad), parte III (datos preclínicos) y parte IV (datos clínicos).
El volumen 2C fue publicado en el año 2000 como consecuencia de la necesidad de disponer de guías regulatorias específicas.
La siguiente actualización del Volumen 2B tuvo en cuenta los acuerdos internacionales sobre la estructura y formato común del Documento Técnico Común (CTD), acordado en noviembre del 2000, dentro de la Conferencia Internacional de Armonización (ICH).
Se aprobó entonces el CTD, actual estructura de carpetas de los dosieres, formato acordado internacionalmente para la preparación de solicitudes y su presentación a las autoridades reguladoras de las tres regiones de ICH de Europa, Estados Unidos y Japón. Esta estructura es obligatoria para la presentación de los dosieres desde 2003.
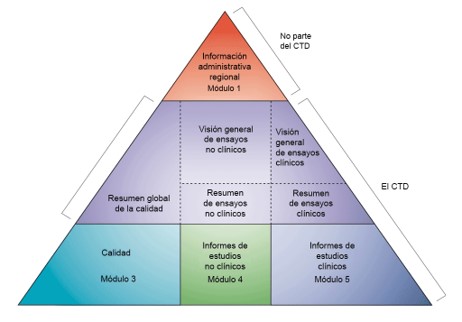
Pero el CTD no nos da información sobre el contenido del expediente y no indica qué estudios y datos son necesarios para la autorización de este expediente. De hecho, el contenido del expediente no es necesariamente idéntico en las tres regiones, y las guías específicas para la región de Europa las encontramos en el EU CTD NTA, donde para cada módulo existe una lista de guidelines relevantes que deben ser tenidas en cuenta en la preparación del dosier. Estas guidelines, además, están en continua revisión.
Por último, desde 2009 es obligatorio la presentación electrónica de los dosieres en la mayoría de los países de Europa. Y es la estructura electrónica de este último formato, lo que nos permite tener, en un único documento, el ciclo de vida de nuestros productos.
Situación actual
Pero a pesar de todos estos cambios y actualizaciones legislativas, hoy en día, podemos encontrarnos en los archivos de nuestra compañía, dosieres completamente desactualizados.


A esta situación, debemos añadir que los dosieres han ido sufriendo modificaciones a lo largo de su ciclo de vida. Estas variaciones se realizaban primero en papel, archivándose en carpetas físicas, y después en los dispositivos electrónicos que fueron apareciendo (disquetes, CDs, las carpetas de nuestro ordenador, etc.)
Pero ¿que más ha contribuido a que nuestros dosieres no estén actualizados?
Principalmente, el desconocimiento de la importancia de los asuntos regulatorios por el resto de los departamentos de la compañía y por tanto falta de comunicación entre los departamentos de registros y las plantas de fabricación.
Debemos ser conscientes que las plantas de fabricación han ido modificando sus procesos de fabricación y control de acuerdo al estado del arte y estos cambios en la mayoría de las ocasiones no se llegaron a remitir a los titulares de comercialización, por tanto, nunca fueron comunicados a las autoridades ni la nueva documentación quedó registrada en los correspondientes dosieres.
Como resultado de lo anteriormente expuesto, tenemos medicamentos que a pesar de saber que están fabricados de acuerdo con la normativa actual y cumplen todos los requisitos de calidad, disponen de una documentación escasa y antigua, lo que no nos permite demostrar ese cumplimiento.
¿Y cuáles son los gaps más comunes que podemos encontrarnos?
- Métodos de análisis obsoletos y/o no validados
- Ausencia del perfil de impurezas
- Declaración de impurezas totales, pero no individuales
- No hay métodos de cuantificación de impurezas
- Metales pesados en lugar del análisis de acuerdo ICH Q3D
- Falta de especificaciones a la caducidad del producto
- Desactualizaciones respecto a las Farmacopeas
- Procesos de fabricación obsoletos
- Fabricantes incluidos en los dosieres que ya no utilizamos
- Documentación de excipientes que no está de acuerdo a la normativa vigente
- Estándares de referencia no actualizados
- Material de acondicionamiento que no cumple con los requisitos legislativos.
El dosier de registro es propiedad de los TAC, pero una de sus obligaciones es enviar la parte de calidad al fabricante y entre ambos deben mantenerlo actualizado.
El TAC presenta el dosier a evaluación, la AEMPS lo autoriza y el fabricante debe fabricar siguiendo estrictamente el proceso aprobado.
La Agencia Española del Medicamento y Productos Sanitarios (AEMPS), en las inspecciones GMP que realiza a los fabricantes de producto terminado, desde hace unos años está revisando los dosieres que estos fabricantes reciben de los TAC y que se supone deben estar actualizados. Sin embargo, la AEMPS ha puesto de manifiesto que esto no es así y que los dosieres localizados en las instalaciones de los fabricantes están muy desactualizados.
¿Cómo podemos ayudarte desde el departamento de Qualipharma Regulatory Affairs a actualizar tus dosieres?
El primer paso es analizar la situación global: de cuántos dosieres hablamos, cómo son de antiguos y seguidamente establecemos una estrategia de trabajo.

Ejemplo de estrategia de trabajo:
- Analizar la documentación disponible del dosier:
- Dosier original (si existe)
- Documentación de las variaciones: en papel, soporte electrónico …
- Recopilar todas las autorizaciones, desde la primera autorización del dosier hasta la última variación presentadas para crear un histórico del producto.
- Revisión de la documentación recopilada hasta el momento y evaluar su validez de acuerdo a la legislación actual.
- Recopilar los documentos generados por el fabricante: guías de fabricación y acondicionado, especificaciones de todos los materiales, métodos de control, datos de estabilidad, etc.
- Comparar la documentación recibida por los fabricantes con la documentación identificada como autorizada y vigente.
- Identificar las diferencias y definir una estrategia de variaciones para actualizar el dosier
- Una vez autorizadas todas las variaciones, se prepara un dosier consolidado en formato eCTD. Este dosier consolidado será la secuencia 0000 y a partir de aquí ya dispondremos de toda la documentación actualizada.
Como conclusión, señalar que es importante, que al final de este trabajo tengamos identificados y archivados los siguientes documentos:
- Autorización inicial
- Ultimo anexo II
- Textos y maquetas autorizadas
Más información en nuestra sección sobre Regulatory Affairs
Evita sorpresas al adquirir con la autorización y comercialización de medicamentos
Validaciones, cualificaciones, requisitos FDA, formación. Tenemos los servicios que se adaptan a tus necesidades.