
En esta cuarta entrega de los artículos en los que desgranamos el nuevo Anexo 1, nos centraremos en las nuevas tecnologías de producción en ambientes estériles, de las que destacan la técnica de producción de ampollas en continuo “Blow-Fill-Seal” (BFS)
(Soplado-Llenado-Sellado) y la Liofilización. Adicionalmente, y no menos importante, analizaremos los cambios que afectan al área de Control de Calidad.
Dados los requisitos de la fabricación estéril, al tener que trabajar en ambientes prácticamente libres de partículas, nos vemos obligados a recurrir a ambientes de Grado A en donde poder llevar a cabo las operaciones de alto riesgo, como por ejemplo las operaciones de llenado.
En este punto la tecnología BFS juega un papel importante para facilitar una producción de ampollas monodosis estériles y libres de partículas.
Blow-Fill-Seal (BFS)
Las unidades de Blow-Fill-Seal están específicamente diseñadas para que, de forma continua, se formen, se llenen y se sellen ampollas, botellas o frascos de manera automática.
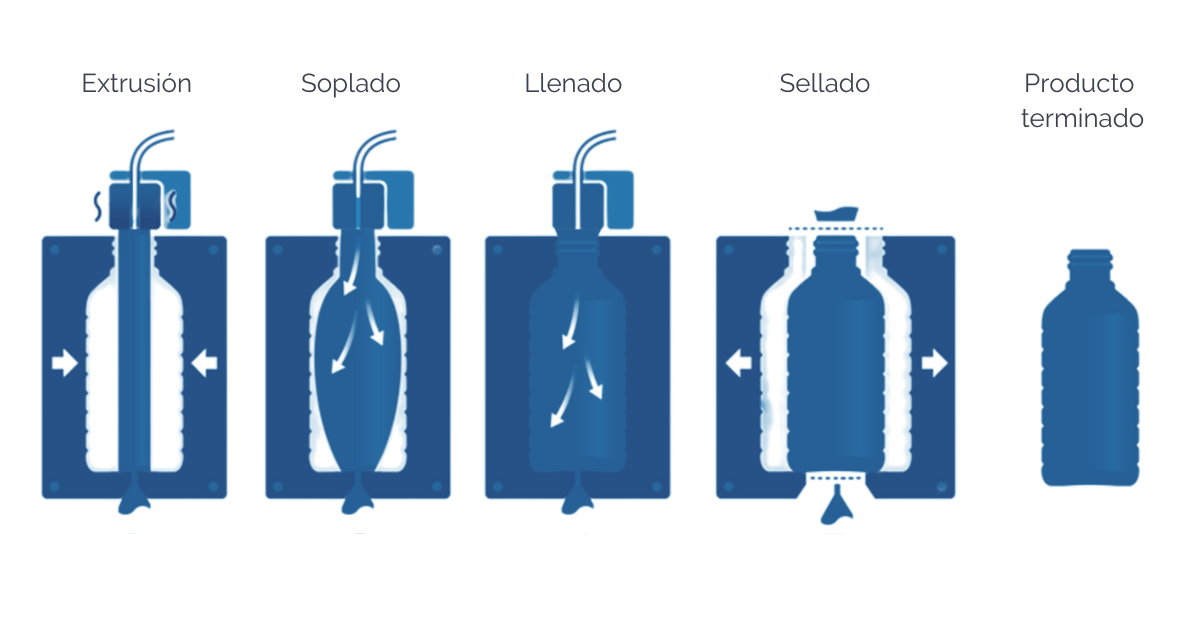 El aire en contacto con las superficies criticas debe someterse a una filtración adecuada, de este modo un equipo de BFS que esté provisto de una fuente de aire de grado A, puede instalarse un entorno de grado D si el proceso cuenta con esterilización terminal, siempre que se utilice vestimenta de grado A/B. El proceso de llenado puede llevarse a cabo en entornos de Grado C salvo en los casos en que exista un riesgo inusual en cuyo caso el llenado deberá llevarse cabo en Grado A, en un entorno, al menos, de Grado C.
El aire en contacto con las superficies criticas debe someterse a una filtración adecuada, de este modo un equipo de BFS que esté provisto de una fuente de aire de grado A, puede instalarse un entorno de grado D si el proceso cuenta con esterilización terminal, siempre que se utilice vestimenta de grado A/B. El proceso de llenado puede llevarse a cabo en entornos de Grado C salvo en los casos en que exista un riesgo inusual en cuyo caso el llenado deberá llevarse cabo en Grado A, en un entorno, al menos, de Grado C.
También se deben considerar otros controles para el aseguramiento de la esterilidad de los productos como la monitorización de los parámetros críticos y de las alarmas durante cada lote además de los test de integridad de filtro.
El control del entorno y el programa de monitorización deberán tener en consideración el movimiento de partículas y los patrones de flujo de aire generados durante un proceso BFS y la afectación sobre los puntos calientes del proceso, por ejemplo, realizando estudios de movimiento con humo o similares.
La monitorización del entorno debe llevarse a cabo evaluando elementos como la configuración del sistema, prestando especial atención en los movimientos operacionales de la máquina y los patrones de aire generados durante el proceso sin olvidarnos del sistema de filtración de aire y su integridad, la integridad del sistema de refrigeración y el diseño de los equipos y la instalación. Todo ello sumado a un apropiado control de partículas y contaminación microbiológica del material de partida de las ampollas y a un sistema de extrusión capaz de ofrecer un aseguramiento de la esterilidad del moldeado que deberá entenderse completamente y validarse. La frecuencia de muestreo, el bioburden y los niveles de endotoxinas (cuando aplique) del material de partida deberán definirse dentro de la estrategia de control de la contaminación o Contamination Control Strategy (CCS).
Los moldes usados para la formación de las ampollas son considerados elementos críticos y cualquier cambio o modificación deberá contar con un aseguramiento de la integridad del contenedor del producto final que deberá estar respaldada por la pertinente validación.

En resumen, el empleo de maquinaria BFS garantiza un sistema de producción de envases unidosis sin apenas intervención humana, situación que facilita el cumplimiento de las características de los productos estériles.
Vamos ahora a ver en detalle otra de las técnicas de producción más interesantes, desde el punto de vista tecnológico, para la producción de envases de productos estériles.
Liofilización
La liofilización es el método de elección para los productos destinados a la administración parenteral al ser un método que ayuda a la conservación de los productos una absoluta esterilidad y garantiza una rápida reconstitución del producto seco en el punto de uso.
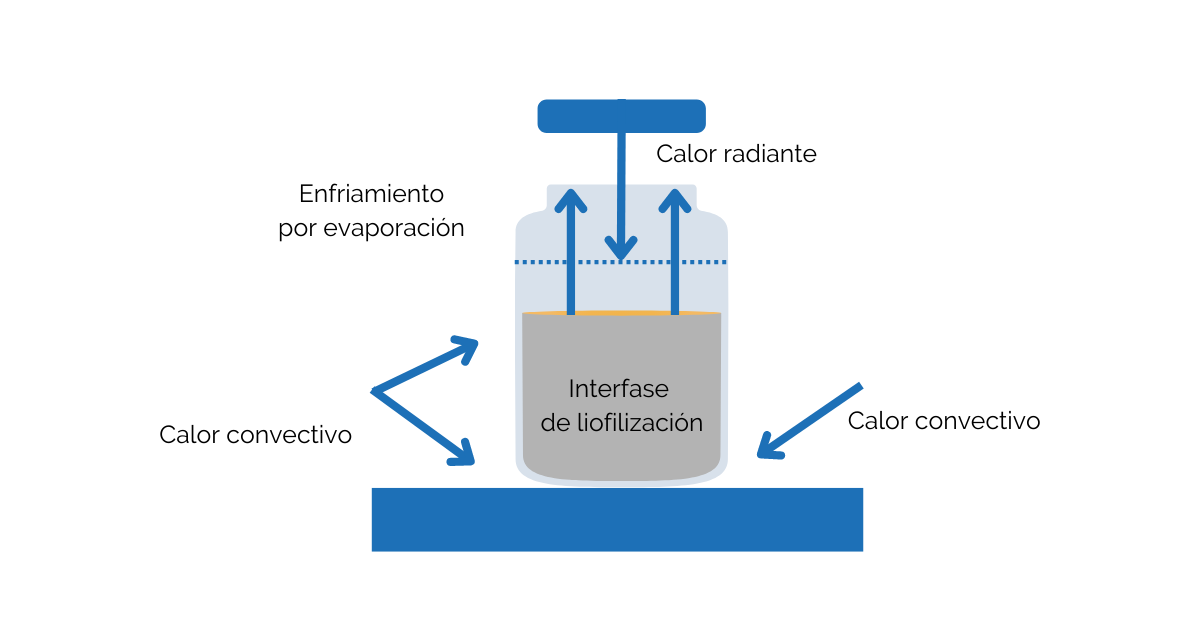 Al tratarse de una etapa crítica y como cualquier otra actividad que pueda afectar a la esterilidad del producto, el equipo de liofilización y sus procesos deben estar diseñados para asegurar que la esterilidad del producto y los materiales se mantenga durante todo el proceso, previniendo la contaminación microbiológica y por partículas desde la etapa de llenado hasta la finalización del proceso de liofilización. Todas las medidas de control y su evaluación deben determinarse en el CCS de la planta.
Al tratarse de una etapa crítica y como cualquier otra actividad que pueda afectar a la esterilidad del producto, el equipo de liofilización y sus procesos deben estar diseñados para asegurar que la esterilidad del producto y los materiales se mantenga durante todo el proceso, previniendo la contaminación microbiológica y por partículas desde la etapa de llenado hasta la finalización del proceso de liofilización. Todas las medidas de control y su evaluación deben determinarse en el CCS de la planta.
La esterilización del liofilizador, que deberá ser llevada a cabo de forma regular acorde al diseño del sistema y la del equipamiento asociado (bandejas, soportes para los viales, etc.) deberá estar validada y los tiempos de espera entre los ciclos de esterilización deberán estar debidamente establecidos y comprobados durante la Simulación del Proceso Aséptico (APS).
Es importante destacar que los liofilizadores de carga/descarga manual deberán esterilizarse antes de cada carga y en aquellos equipos en los que la intervención humana queda excluida (equipos dotados de sistema de carga/descarga automáticos) la frecuencia de esterilización deberá estar justificada y documentada como parte del CCS.

La esterilidad del liofilizador deberá mantenerse tras la esterilización y durante el proceso. El filtro utilizado para el mantenimiento de la esterilidad deberá esterilizarse antes de cada uso y los resultados de su test de integridad deberán formar parte de la documentación de certificación del lote. Por otro lado, no podemos olvidarnos de la cámara del liofilizador cuya integridad del vacío/fugas deberá estar documentada y el valor máximo de fugas admisibles deberá estar especificado y verificado al inicio de cada ciclo de liofilización. Todos estos controles y test se complementarán con una revisión periódica de la integridad de las bandejas para asegurar que no haya sufrido deformaciones o daños.
Todas las operaciones llevadas a cabo antes de la liofilización (carga del liofilizador, manipulación de viales) deberán realizarse bajo un ambiente de Grado A y estar diseñadas para minimizar la intervención humana directa. Alternativamente y siempre que una validación lo respalde, se podrán utilizar contenedores cerrados en ambientes Grado A y no reabiertos en Grado B para proteger viales parcialmente cerrados. Recordemos que los viales no sellados deberán permanecer en ambientes de Grado A y estar separados del personal mediante una tecnología de barrera física u otras medidas adecuadas.
Otros sistemas
Como acabamos de ver, los equipamientos BFS y la liofilización son dos herramientas muy interesantes y potentes para obtener presentaciones estériles, pero no son las únicas. Hagamos un repaso a dos de los sistemas que podemos utilizar en la fabricación estéril.
Por un lado, tenemos los sistemas cerrados, que pueden ser elementos de un solo uso (sistemas desechables) o fijos (recipientes con conducciones fijas). El uso de este tipo de sistemas puede reducir el riesgo de contaminación ya que están diseñados para reducir la complejidad de manejo o la intervención humana y asegurar el mantenimiento de la esterilidad. En el caso que uno de estos sistema debiera ser abierto, la apertura deberá realizarse en entornos clasificados, Grado C para procesos con esterilización terminal o Grado A para proceso asépticos o deberán ser sometidos a posteriores limpiezas y desinfecciones (esterilizaciones en el caso de procesos asépticos).
Aunque pudiera parecer lo contrario, los sistemas de un solo uso o Single Use System (SUS) no están exentos de riesgos. Los riesgos específicos de los SUS deberán ser evaluados como parte del CCS. La verificación de la esterilidad de los SUS deberá llevarse a cabo como parte de la evaluación del proveedor y en la recepción y uso de cada unidad. Además, la adsorción y reactividad del producto con las superficie del SUS deberán evaluarse bajo condiciones de proceso.
Los SUS deberán estar diseñados para que puedan mantener su integridad estructural a lo largo de todo el proceso productivo, especialmente cuando puedan verse expuestos a condiciones más extremas, durante procedimientos rutinarios o durante su traslado.
Control de Calidad
La sección de Control de Calidad del Anexo 1 incluye, como primer punto a cumplir, que se debe contar con personal debidamente formado y con experiencia en microbiología, ya que debe brindar apoyo en el diseño del proceso de fabricación, en el control ambiental o en cualquier investigación en la que se deba evaluar el impacto de los eventos sucedidos con la microbiología en la seguridad del producto estéril.
También indica que debe haber un mayor control sobre las materias primas y/o componentes con la finalidad de minimizar la contaminación microbiológica; por lo que las especificaciones de cada material de partida deberá incluir requisitos de calidad microbiológica cuando haya sido definida por los controles de monitorización y/o por la Estrategia de Control de Contaminación o Contamination Control Strategy (CCS).
El uso de la Estrategia de Control de Contaminación (CCS) también deberá aplicarse para aquellos productos sobre los que no se pueda realizar el ensayo de esterilidad antes de su liberación, porque la vida útil del producto sea muy corta. En dicha estrategia deberán identificarse los riesgos, y establecer la monitorización necesaria para mitigar los riesgos detectados.
El sistema de liberación paramétrica se sigue permitiendo para la liberación de lotes, siempre y cuando se considere el ensayo de carga biológica o Bioburden previo a la esterilización final como una prueba dentro del proceso y, por tanto, se analice en cada lote fabricado, tanto para el producto llenado asépticamente como los productos esterilizados terminalmente. En aquellos casos que aplique, el nivel de pirógenos (endotoxinas) también deberá controlarse como un ensayo adicional. Los resultados deberán considerarse como parte de la revisión final del lote.
Adicionalmente, deberá demostrarse que los procesos de esterilización utilizados para descontaminar las superficies externas de las muestras de esterilidad, como por ejemplo peróxido de hidrógeno vaporizado o ultravioleta, no tendrán impacto negativo sobre la sensibilidad del método analítico utilizado.
- Ensayo de Esterilidad:
El ensayo de esterilidad en el producto terminado únicamente debe considerarse como el último elemento de una serie de medidas de control mediante las cuales se asegura la esterilidad, asimismo dicho ensayo deberá efectuarse en condiciones asépticas, que sean como mínimo iguales la misma clasificación de sala limpia.
Adicionalmente, se introduce la evaluación de riesgo mediante la selección del Peor Caso o Worst Case para establecer los puntos del lote donde haya un mayor riesgo de contaminación, seguidamente definir el número de muestras a tomar y la ubicación de estas. A la par, indica que cualquier organismo encontrado durante la prueba de Bioburden, se debe identificar y evaluar su impacto sobre la efectividad en el proceso de esterilización.
- Toma de Muestras:
La toma de muestra debe ser representativa de todo el lote y como se indicó anteriormente, se deben tomar en los puntos del lote donde se considere que existe un mayor riesgo de contaminación, bajo la evaluación del peor caso, entre los ejemplos que se mencionan con una mayor fuente de contaminación tenemos:
– En el caso de los productos que se hayan llenado asépticamente, las muestras deberán incluir envases llenos del inicio, mitad y final del lote.
– Después de cualquier intervención significativa (por ejemplo, intervenciones en las que se haya violado la integridad de una barrera (puerta abierta)) o una intervención del operador en zonas críticas.
– En el caso de los productos que han sido esterilizados por calor en sus envases finales, las muestras tomadas deben ser de la parte potencialmente más fría o la más lenta en calentar cada carga.
– Del proceso de liofilización las muestras deberán ser tomadas de diferentes cargas.
En este punto es importante indicar que el borrador del Anexo 1 hace la salvedad de que, si en el proceso de fabricación da lugar a sub-lotes (por ejemplo, diferentes cargas de liofilización), deberán tomarse muestras para el ensayo de esterilidad por separado y se debería considerar la posibilidad de realizar otros ensayos de producto terminado también separados
- Monitorización Ambiental:
La monitorización ambiental, resultados y tendencias, deberán ser revisados como parte de la certificación de los lotes, teniéndose que disponer de planes por escrito que describan las acciones que se deben tomar cuando los resultados de la monitorización ambiental se encuentren fuera de especificaciones (OOS) o fuera de tendencia (OOT). Para el caso de los productos con vida útil corta, deberá indicarse que la certificación de los lotes se realizará con los datos de monitorización que se tengan disponibles y de entre ellos los más recientes. Para ello se considerará el uso de sistemas de monitorización rápida; dichos métodos deberán estar validados para el producto o proceso en cuestión antes de utilizar los resultados obtenidos en la certificación de los lotes.
En este punto es importante indicar que los planes de monitorización ambiental no solo tendrán que estar definidos para las salas de producción clasificadas como grado A/B sino que en esta nueva edición del anexo 1 las zonas de producción clasificadas como C/D también tendrán que definir un plan de acciones que deba tomar en cuenta lo indicado anteriormente.
Para culminar, podemos resumir que el Anexo 1 se está adaptando a las tecnologías existentes en el mercado que no estaban contempladas en la normativa actual. Dichas tecnologías de producción en ambientes estériles, BFS y liofilización, son dos herramientas muy interesantes y potentes para obtener presentaciones estériles, pero no son las únicas. Existen sistemas cerrados, que pueden ser elementos de un solo uso (sistemas desechables) o fijos (recipientes con conducciones fijas) que también pueden evaluarse para ser utilizadas en la fabricación de productos estériles; siendo lo principal en este punto es que sin importar la tecnología que se aplique lo importante es cumplir con los nuevos límites microbiológicos y de partículas “en reposo” y “en funcionamiento” que se definen en el nuevo anexo.
Por otro lado, se puede decir que en la sección de Control de Calidad figura información más ampliada sobre los “ensayos microbiológicos” que se deben aplicar a los lotes de producto terminado. Estos ensayos abarcan desde la evaluación de los materiales de partida, pasando por la valoración de realizar ensayos de cargas microbiana en productos intermedios, hasta una prueba de Bioburden realizada inmediatamente antes del filtro de esterilización (para productos llenados asépticamente) o antes de la esterilización final.
Adicionalmente, aunque no se discuten en detalle todas las limitaciones de la prueba de esterilidad, las nuevas medidas implementadas en el Anexo 1 permitirán garantizar la esterilidad de los productos ya que se efectuarán más controles, sobre materiales de partida, productos intermedios y condiciones ambientales que permitirán minimizar riesgos de contaminación microbiana en el producto terminado.
Os invitamos a leer el próximo y último artículo de esta secuencia en la que hemos identificado los cambios del nuevo Anexo 1 y sus implicaciones para la industria.
Garantiza tu producto liofilizado
Desde Qualipharma te damos el soporte global de liofilización que necesitas a lo largo de todo el ciclo de vida del producto.
