
The registration of medical products with the United States Food and Drug Administration (FDA) is a crucial step for companies seeking to market their products in one of the largest and most regulated medical device markets in the world. However, the process can be complex, especially for those unfamiliar with industry regulations. For this reason, and to avoid delays and errors that could hinder market access, it is essential to approach the registration process with a clear strategy and a deep understanding of the specific requirements.
Classification of Medical Devices
The FDA classifies medical devices into three categories based on the level of risk they pose to patients and users. This classification determines the applicable regulatory requirements, including the appropriate registration pathway and the necessary controls for marketing the device in the United States.
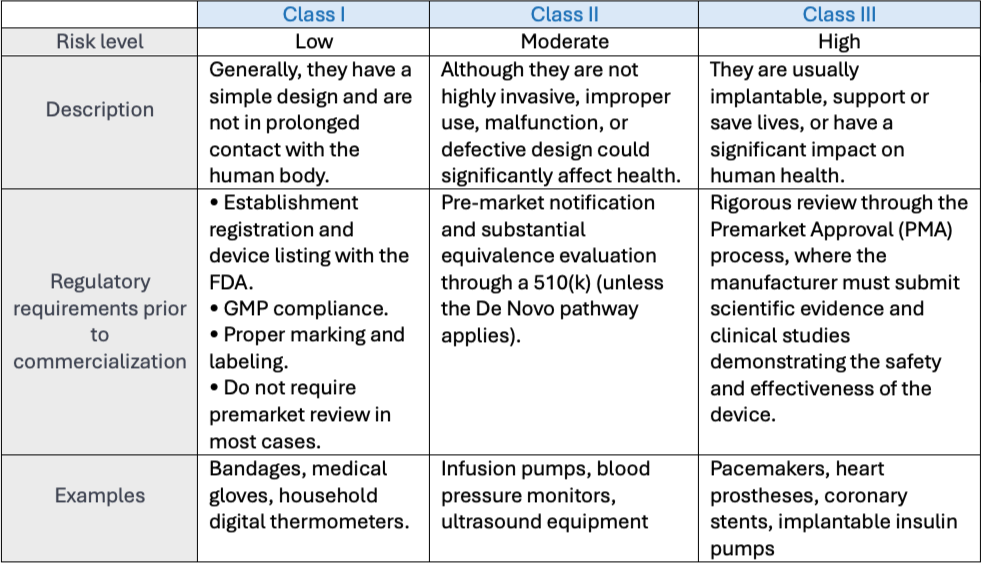
Correctly determining the classification of a medical device is crucial to defining the appropriate regulatory strategy. An incorrect classification can lead to approval delays, unnecessary costs, or even prevent the product from being marketed in the U.S.
To confirm the classification of a device, the FDA provides the Product Classification Database, where similar devices can be searched to verify the applicable requirements.
How to Register a Medical Device with the FDA?
This registration follows a structured process that requires detailed attention at each stage:
1. Classification of the Medical Device:
The first step is to determine the device’s class (Class I, II, or III), which defines the associated risk level and the applicable regulatory requirements.
2. Regulatory Strategy:
Once the device is classified, and to ensure the success of its registration, it is vital to establish the correct regulatory strategy, as each product will apply for a specific registration pathway based on its characteristics. The following registration pathways can be found:
• Self-registration (Some Class I and Class II devices)
• Pre-marketing notification (510(k)) (Some Class I and Class II devices)
• De Novo (Some Class I and II devices without a predicate device)
• Premarket Approval (PMA) (Class III devices)
3. Identification of a Predicate Device (if applicable):
For Class II devices and certain Class I devices, it may be necessary to demonstrate substantial equivalence to an already approved predicate device through a Premarket Notification (510(k)).
4. Compliance with Specific Regulatory Standards:
Regardless of the class, all medical devices are subject to the Quality System Regulation (21 CFR 820), including current Good Manufacturing Practices (GMP), unless an exception or exemption is specified in 21 CFR 820.
5. Preparation of Technical Documentation:
A complete list of documents must be compiled, including technical descriptions, safety and efficacy test reports, and clinical data if applicable.
6. Submission of the Application to the FDA:
Once the Technical Documentation is prepared, it is submitted to the FDA through the legally established pathway.
7. Follow-up and Response to FDA Requests:
The FDA may request additional information or clarifications, so efficient follow-up is key to avoid delays.
Don’t miss the second part of this blog, where we will explore in detail each registration pathway and its specific requirements.