
Como anticipábamos en el anterior blog, las empresas que tienen un medicamento autorizado en uno de los Estados miembros de la UE pueden solicitar que se reconozca esta autorización en otros países de la Unión. Este proceso permite a cada Estado miembro basarse en las evaluaciones científicas de los demás.
El procedimiento de reconocimiento mutuo (MRP) puede iniciarse para obtener autorizaciones de comercialización en varios Estados miembros cuando el medicamento en cuestión ya haya recibido una autorización de comercialización previa en cualquier Estado miembro en el momento de la solicitud.
El procedimiento de reconocimiento mutuo se divide en los siguientes pasos:
- Revisión y validación por parte de la autoridad competente que actuó como Estado miembro de referencia durante la evaluación del dossier en su presentación original.
- Emisión del informe de evaluación actualizado por parte del Estado miembro de referencia (90 días).
- Validación por parte de los nuevos Estados miembros interesados en formar parte del procedimiento.
- Desarrollo de la evaluación por parte de los Estados miembros interesados (90 días).
- Debate en el grupo de coordinación, si es necesario.
- Etapa de la autorización nacional de comercialización.
En el caso de una solicitud de genérico, el solicitante deberá considerar cuidadosamente la elección del producto de referencia para su producto y deberá discutir esta elección con el Estado miembro de referencia. El solicitante tiene que demostrar que su medicamento es bioequivalente con el medicamento de referencia. Los Estados miembros concernidos aceptarán la demostración de la bioequivalencia, independientemente de que el medicamento de referencia haya sido autorizado o no en todos los Estados miembros afectados. La autoridad competente del Estado miembro en el que el medicamento de referencia tiene o ha tenido una autorización de comercialización, transmitirá, a petición del Estado miembro de referencia, en el plazo de un mes, una confirmación de que el medicamento de referencia está o ha sido autorizado, junto con la composición completa del medicamento de referencia y, en su caso, otros datos.
El titular de la autorización de comercialización (TAC) debe asegurarse que:
- El producto se considerará un medicamento en todos los Estados miembros afectados.
- El producto entra en el ámbito de aplicación del procedimiento de reconocimiento mutuo.
- Los módulos 3, 4 y 5 están debidamente actualizados o las indicaciones clínicas buscadas han sido previamente autorizadas para un medicamento que contiene la misma sustancia activa en los Estado(s) miembro(s) en cuestión.
- Poder respaldar las indicaciones del resumen de las características del producto, el prospecto y el etiquetado.
- Que los expedientes en el Estado miembro de referencia y en los Estados miembros afectados son los mismos;
- Las modificaciones o renovaciones de la autorización original han sido autorizadas por el Estado miembro de referencia antes de iniciar el procedimiento;
- El texto final del resumen de las características del producto aprobado, prospecto y el etiquetado, deben estar disponibles en la lengua nacional del Estado miembro de referencia, con las traducciones apropiadas y teniendo en cuenta las directrices pertinentes.
Podríamos resumir el procedimiento en una línea de tiempo de la siguiente manera:
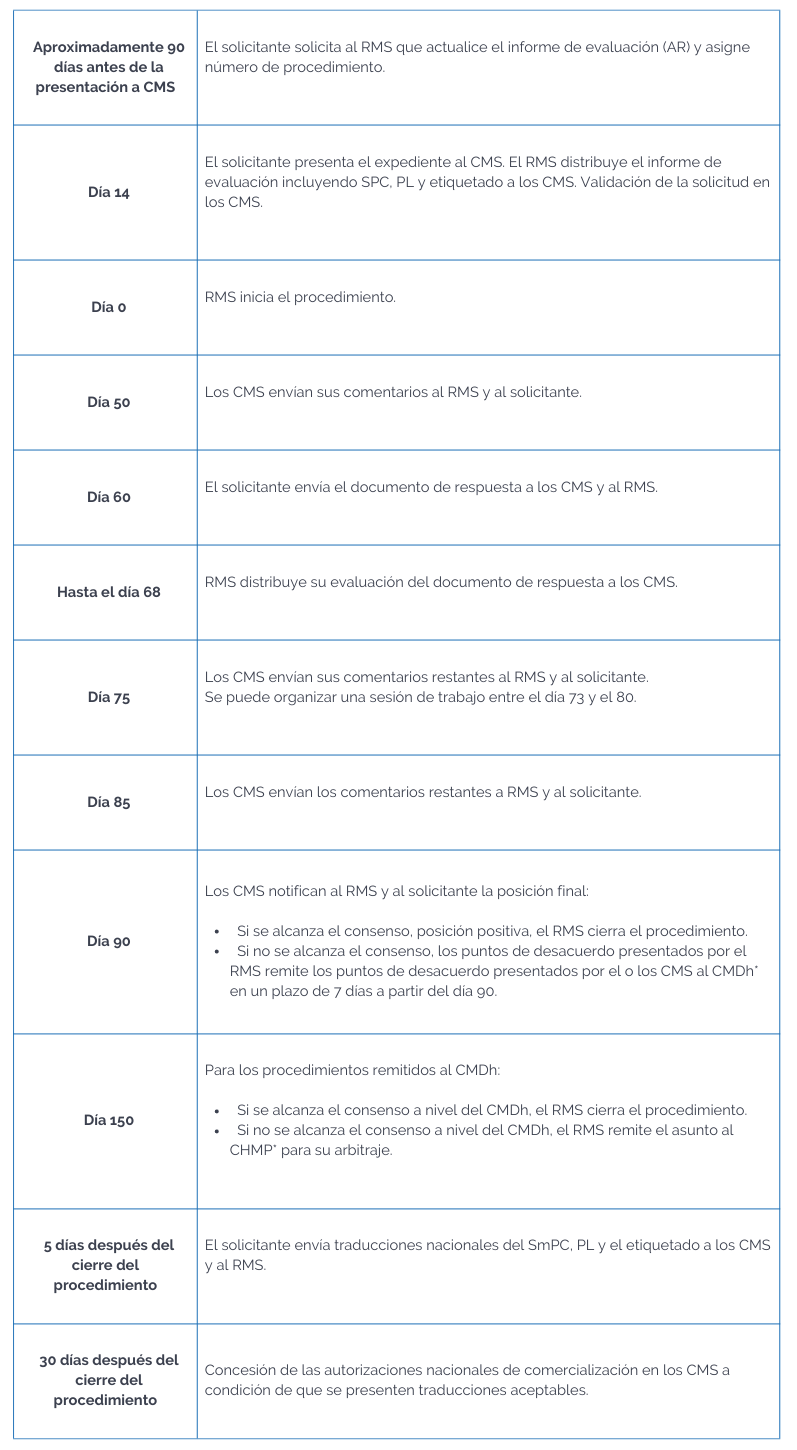
* Committee for Medicinal Products for Human Use (CHMP).
Coordination Group for Mutual Recognition and Decentralised Procedures – Human (CMDh).
Conoce la legislación europea
En Qualipharma te ayudamos a definir la estrategia regulatoria más adecuada para poner tu producto en el mercado